La Sindrome di Morris Caso Clinico #10
Caterina è una bellissima ragazzina di 15 anni, alta, longilinea, sorridente.
Viene accompagnata presso il mio ambulatorio dalla mamma, preoccupata perché Caterina non ha ancora avuto la sua prima mestruazione.
Procedo con la visita e noto subito che la vagina non presenta alcun pelo pubico, allo stesso modo anche le ascelle non hanno peluria. La pelle del viso è liscia, senza acne, il seno è sviluppato normalmente.
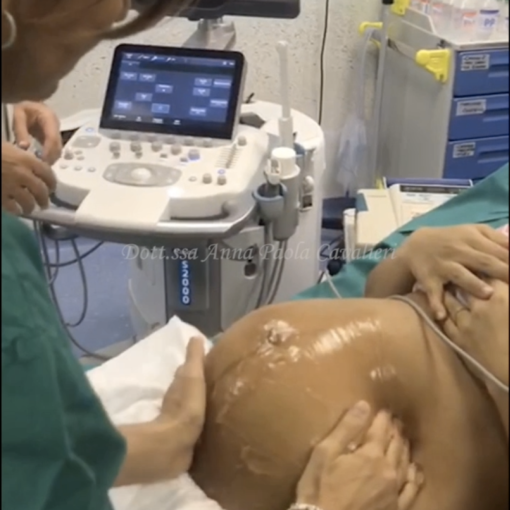
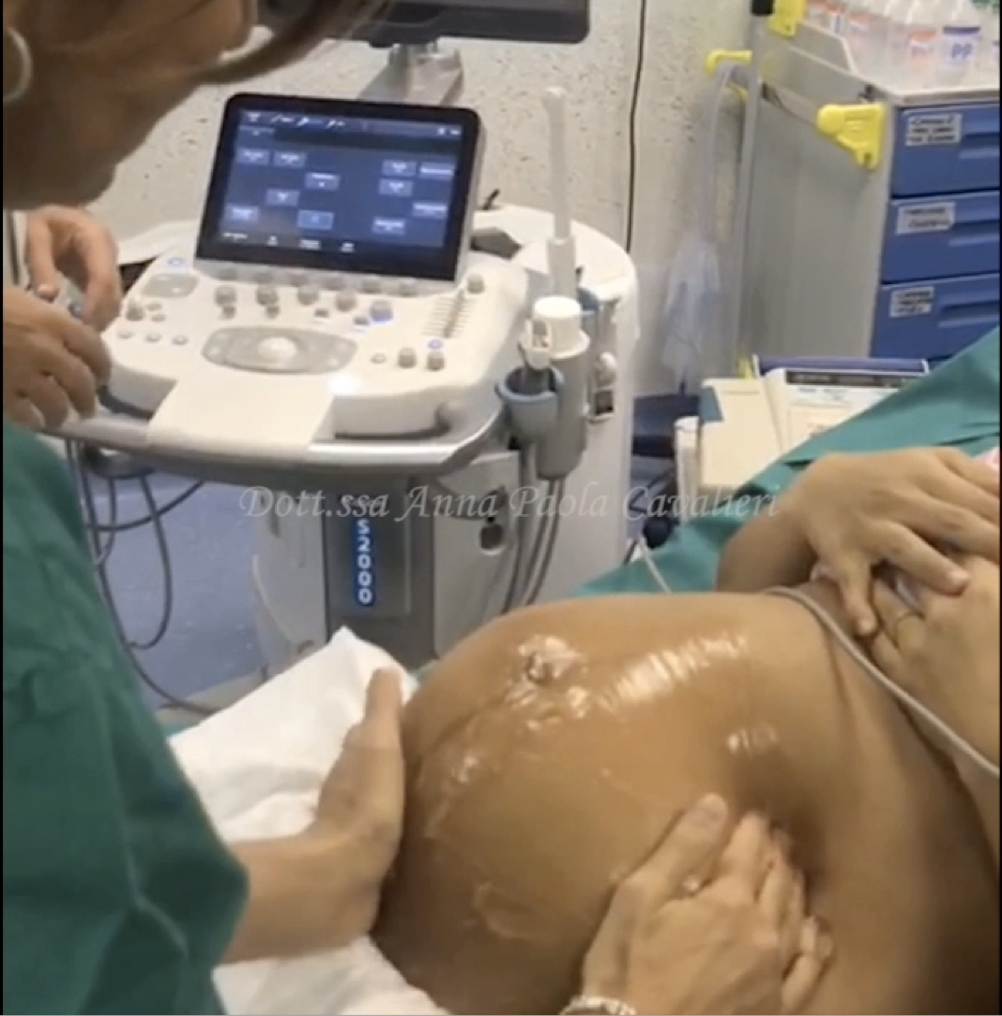
Procedo con una ecografia pelvica addominale e non riesco a vedere l’utero né le ovaie.
Richiedo a questo punto una serie di esami del sangue tra cui il cariotipo, cioè l’analisi dei cromosomi.
Ricevuti i risultati degli esami la diagnosi è fatta. Caterina presenta un cariotipo maschile, 46XY.
Una diagnosi molto difficile da comunicare… sindrome di Morris.
La sindrome di Morris o sindrome da insensibilità agli androgeni è una rara patologia dello sviluppo sessuale che inizia già in utero.
Le persone affette da tale sindrome sono geneticamente di sesso maschile, con un cromosoma X e un cromosoma Y, ma il loro aspetto è femminile in quanto il loro corpo non è capace di rispondere agli ormoni maschili (androgeni).
La malattia è dovuta da una anomalia genetica che può essere ereditaria (legata al cromosoma X materno) oppure casuale cioè legata ad una mutazione che si verifica spontaneamente.
Tale anomalia rende i recettori delle cellule maschili incapaci di rispondere agli ormoni androgeni o con una risposta limitata. L’ insensibilità agli androgeni può essere quindi parziale o totale.
Quando è totale, come nel caso di Caterina, queste persone appaiono esternamente come donne, con seno sviluppato e genitali esterni femminili, ma mancano gli organi genitali interni femminili (utero e ovaie), quindi non avranno mestruazioni ne’ potranno avere gravidanze.
Questi pazienti crescono come donne e sviluppano una identità di genere femminile.

Non hanno però genitali interni femminili bensì maschili, in particolare i testicoli sono localizzati all’interno dell’addome o della pelvi e andranno rimossi chirurgicamente in quanto esiste la possibilità che sviluppino forme cancerose.
Di solito la peluria pubica e ascellare è scarsa, l’acne assente.
Nelle forme parziali di insensibilità agli estrogeni (sindrome di Reifenstein) il quadro clinico varia da genitali esterni di aspetto femminile a quadri misti o maschili.
Anche l’aspetto esterno può variare da caratteristiche prevalentemente femminili a quadri con aspetto maschile. Anche in questi ultimi casi però i pazienti risultano molto spesso sterili e frequentemente si osserva uno sviluppo anomalo del seno alla pubertà.
La sindrome di Morris in forma completa ha una frequenza di circa 2-5 individui ogni 100000 persone di sesso genetico maschile, mentre la forma parziale è ancora più rara .
Se l’insensibilità agli ormoni maschili è parziale, i medici sono in grado di sospettare la malattia già dopo la nascita, in quanto i genitali esterni presentano delle caratteristiche sospette. Se invece l’insensibilità è completa il neonato ha, esternamente, tutte le sembianze di una bambina.
In questi casi la diagnosi quindi avviene di solito alla pubertà, poiché non si verificano le mestruazioni ne’ la crescita della peluria ascellare e pubica.


{kind=link}
{kind=link}
Un commento su “La Sindrome di Morris Caso Clinico #10”
Grazie dove lavora privatamente ? A Modena Bologna Ferrara conosce colleghi che si occupano come lei di quello che tratta?